
0
Suivez-nous
Deux scientifiques dont les travaux ont marqué le début de la première thérapie approuvée utilisant l’outil d’édition génétique CRISPR ont remporté le Breakthrough Prize in Life Sciences de 3 millions de dollars.
Les lauréats – Dr Swee Lay Theindu National Heart, Lung and Blood Institute (NHLBI), et Dr Stuart H. Orkinde l’Université Harvard, a partagé le prix pour la recherche fondamentale qui a conduit au développement d’une thérapie génique qui traite les troubles sanguins, la drépanocytose et la bêta-thalassémie.
Le Prix de la percée en sciences de la vie est décerné depuis 2013 pour reconnaître les réalisations dans le domaine des sciences de la vie.
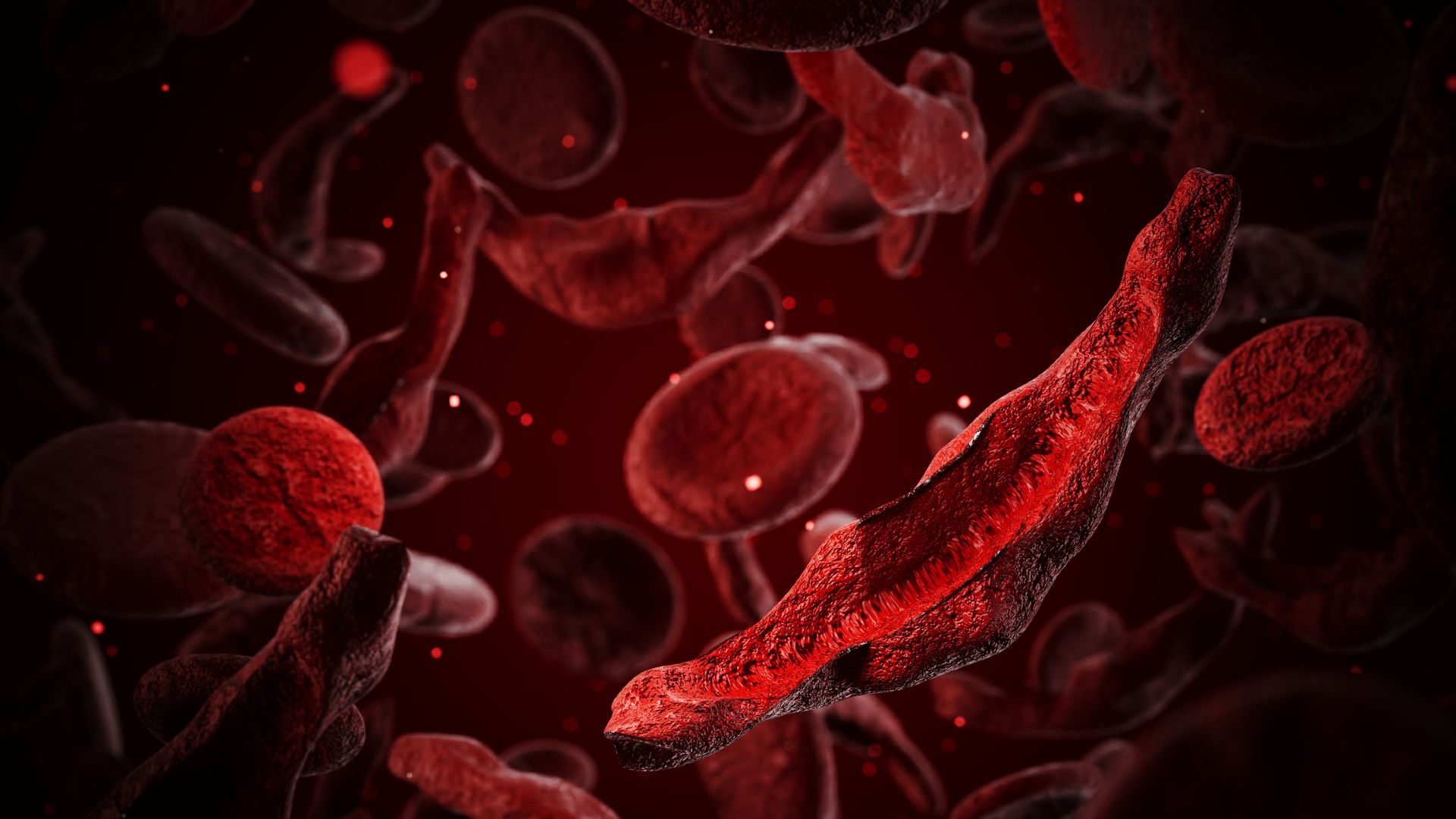
Troubles sanguins mortels
La drépanocytose touche environ 7 à 8 millions de personnes à l’échelle mondiale, principalement en Afrique. Chez les personnes atteintes de cette maladie, les globules rouges prennent une forme caractéristique de croissant car l’hémoglobine, la molécule qui transporte l’oxygène à l’intérieur des cellules, se forme. fibrilles rigides et longues qui déforment les cellules. Ces cellules falciformes se collent les unes aux autres, déclenchant la formation de caillots sanguins, et elles éclatent et meurent facilement, entraînant une faible numération des globules rouges.
Les patients sont souvent confrontés à des épisodes de douleur atroces, appelés « crises », lorsque les globules rouges bloquent les vaisseaux sanguins. Ces blocages peuvent endommager des organes comme les poumons, le foie et la rate. Les blocages dans les poumons peuvent également déclencher un « syndrome thoracique aigu », qui épuise les niveaux d’oxygène et constitue le principale cause de décès chez les patients drépanocytaires.
Dans bêta-thalassémiele corps ne produit pas – ou produit des quantités moindres – une partie de la molécule d’hémoglobine, ce qui signifie que les personnes atteintes de formes graves de la maladie doivent recevoir des transfusions sanguines à vie. Casgevy est approuvé pour traiter cette forme grave de la maladie.
Thein, chercheuse principale au NHLBI, a commencé ses travaux dans les années 1980 en essayant de comprendre pourquoi certaines personnes atteintes de ces troubles présentaient des formes beaucoup plus bénignes de la maladie que d’autres.
La question était apparue des décennies plus tôt, lorsque le Dr Janet Watson, pédiatre basée à New York, avait montré que les nourrissons qui développaient plus tard la drépanocytose ne présentaient aucun symptôme et avaient des globules rouges non drépanocytaires.
Dès que les enfants étaient en bas âge, les symptômes de la maladie sont apparus.
Des travaux de suivi ont montré que les gens produisent différents types d’hémoglobine à différents stades de développement : « l’hémoglobine fœtale » est produite dans l’utérus, et sa production s’arrête à mesure que les bébés grandissent et que « l’hémoglobine adulte » prend le relais.
« J’ai commencé à rassembler des familles – des patients – atteints de thalassémie légère, pour essayer au moins de découvrir la génétique qui se cache derrière », a déclaré Thein à Live Science. « Il semblait évident qu’ils avaient une capacité innée, ou une capacité naturelle, à continuer à produire de l’hémoglobine fœtale. »
Elle a analysé les gènes de plusieurs familles ayant des antécédents de maladie, notamment une famille d’origine indienne qui comprenait plus de 200 membres, s’étendait sur sept générations et vivait sur plusieurs continents.
Réprimer le répresseur
Une idée cruciale est venue d’un étude de paires de jumeaux identiques et fraternels qui produisait des taux d’hémoglobine fœtale très élevés ou très faibles. Cela a permis à Thein et à ses collègues d’identifier variantes génétiques qui ont affecté la production d’hémoglobine fœtale. Ils se sont concentrés sur une région d’un gène sur le chromosome 11 appelée BCL11A.
L’équipe de Thein a découvert que le gène arrête la production d’hémoglobine fœtale à mesure que les bébés grandissent. « C’est un répresseur », a déclaré Thein. Mais lorsque les gens étaient porteurs de certaines versions de BCL11A, le répresseur ne réprimait pas et la production d’hémoglobine fœtale se poursuivait à des niveaux élevés tout au long de la vie.
À partir de là, il n’était pas difficile de conclure que réprimer le répresseur pourrait être une bonne stratégie pour traiter les personnes atteintes de versions sévères de drépanocytose ou de bêta-thalassémie. Les recherches d’Orkin se sont avérées essentielles à cette avancée.
Orkin – qui est hématologue pédiatrique et oncologue au Boston Children’s Hospital, au Dana-Farber Cancer Institute, à la Harvard Medical School et au Howard Hughes Medical Institute – a montré comment le répresseur a médié le passage à l’hémoglobine adulteet que l’édition génétique pourrait cibler la région.
La société de biotechnologie Vertex a ensuite utilisé l’outil d’édition génétique copier-coller CRISPR pour couper la région répresseur de BCL11A.
Ces travaux ont finalement conduit au développement de Casgevy. L’administration de la thérapie consiste à extraire les cellules de la moelle osseuse d’une personne, à modifier le répresseur BCL11A à l’aide de CRISPR, puis à réinjecter les cellules de la moelle osseuse génétiquement modifiées au patient. Les cellules modifiées commencent à produire des globules rouges avec des taux élevés d’hémoglobine fœtale.
Il s’agit du premier « remède fonctionnel » contre la drépanocytose, et il a transformé la vie des rares personnes qui en ont bénéficié. Mais ce n’est pas un remède accessible à toutes les personnes atteintes de la maladie, et il présente certains inconvénients, a déclaré Thein. Le processus de traitement lui-même peut prendre jusqu’à un an, coûte quelques millions de dollars et nécessite une chimiothérapie sévère pour libérer de l’espace dans la moelle osseuse afin que les cellules souches génétiquement modifiées puissent s’enraciner.
« Physiquement, c’est très éprouvant pour le patient », a déclaré Thein.
En outre, la drépanocytose et la bêta-thalassémie touchent principalement les populations d’Afrique, d’Asie et du bassin méditerranéen, où les ressources et les installations nécessaires à un tel traitement peuvent ne pas être disponibles. En conséquence, les scientifiques travaillant sur la thérapie génique se tournent vers une approche « in vivo », qui implique « d’injecter réellement la machinerie d’édition génétique dans le patient », a déclaré Thein. Cela éliminerait le besoin d’extraire, de modifier et de réinjecter les cellules de la moelle osseuse.
En fin de compte, le besoin de davantage de médicaments – y compris des pilules, des injections ou des perfusions moins chères et plus faciles à administrer – est toujours pressant, a déclaré Thein.
Thein a étudié un médicament appelé Mitavipat. Le médicament, qui est actuellement approuvé pour le traitement du déficit en pyruvate kinase, une maladie du sang, et bêta-thalassémiesemble agir en améliorant la santé métabolique globale des globules rouges, a déclaré Thein.
Certains patients prenant ce médicament « suivent ce traitement depuis six ans, et cela a vraiment fait une grande différence », a-t-elle déclaré, mais des tests supplémentaires sont nécessaires pour approuver son utilisation chez les personnes atteintes de drépanocytose.